前言,如何把数据从百度云上下载到linux服务器上
可以直接通过pip下载:pip install bypy -y
第一次使用时需要随便输入一个命令以激活授权界面,如输入 bypy info
然后打开提示的连接
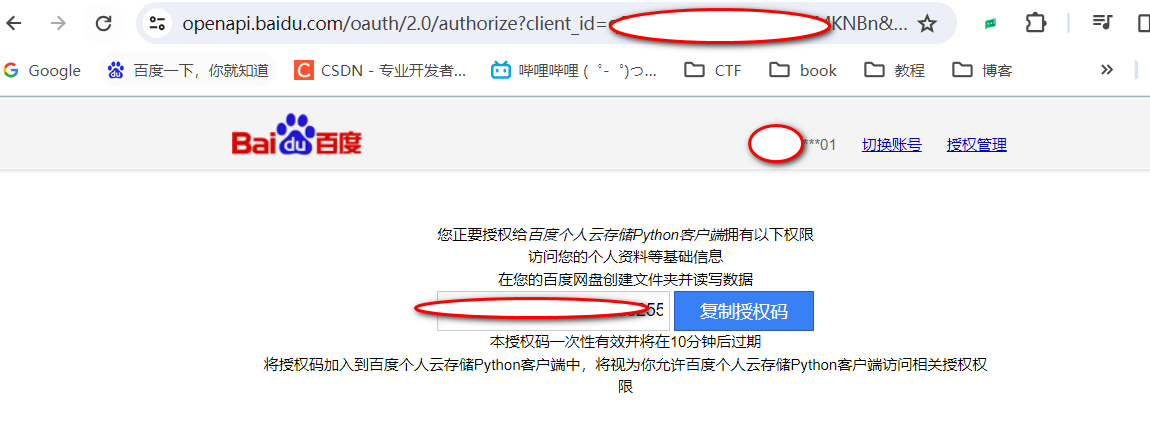
将复制的内容粘贴到终端后回车,等待即可。
登陆成功后会提示如下信息
登录百度网盘(我的应用数据/bypy/你的文件名/*/文件)
有多个文件建议十个放到一个文件夹里,这样下载出错方便排查
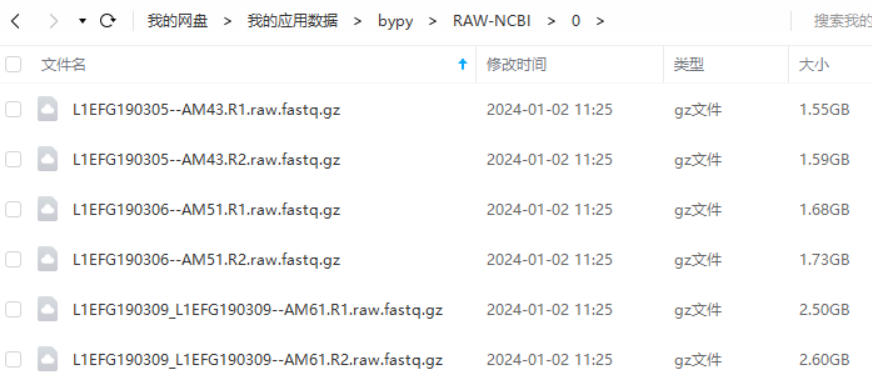
我写了一个脚本可以自动安装download.sh
1 | bypy downdir /RAW-NCBI/0/ ./0/ |
因为我有74个文件,因此分成了7组进行下载
在你想存储的文件夹里输入
1 | nohup bash [download.sh](http://download.sh) > temp.txt & |
(nohup是在Linux中永久运行的命令,&和其他方式均会因为终端退出而中断。>把下载的过程信息存储到temp.txt,&并放在后台运行,这样下载文件的任务就会自动放到后台了)
A.安装
1 | git clone https://github.com/franciscozorrilla/metaGEM.git |
1.找不到env_setup.sh路径
env_setup.sh放在了metaGEM/workflow/scripts/env_setup.sh
但是env_setup.sh里面所有的文件路径是在metaGEM/workflow/下面的,所以要把env_setup.sh复制到metaGEM/workflow/下面运行
详解env_setup.sh文件(提取其中关键部分)(已安装anaconda)
1 | conda create -n mamba mamba -c conda-forge #创建新环境下载maba |
因为conda install 下载速度很慢,所以脚本里使用了mamba方式进行下载。使用方法为下载mamba包,然后在此环境下进行下载,如 mamba install requests
上述文件中mamba env create为创建新环境的语句,-f后面的.yml文件为导出的conda标准环境文件,–prefix 为新创建环境的路径
2.在安装metagem时以下界面卡住
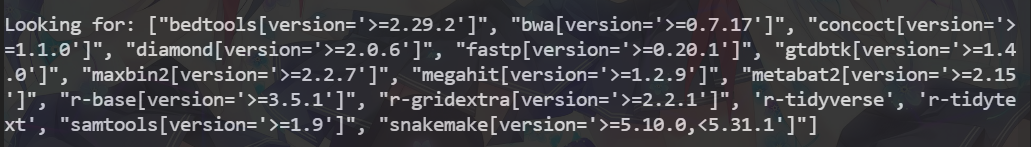
在正常加载时,以下界面至少卡住了20h以上,因此排除网络问题
我们看一下对应的metaGEM_env.yml文件内容
该文件为conda标准创建环境的文件格式
1 | name: metagem |
name指的是创建环境的名称,channels指的是下载通道,其中这个conda-forge是比较重要的一个,其是一个用于托管和发布科学计算、数据分析和机器学习的Python 包的社区项目。在conda-forge通道中,您可以找到为conda构建但尚未成为官方Anaconda发行版一部分的包。有一些Python库不能用简单的conda install安装,因为除非应用conda-forge,否则它们的通道是不可用的。根据我的经验,pip比conda更适合研究不同的通道源。例如,如果你想安装python-constraint,你可以通过pip install来安装,但是要通过cond 来安装。您必须指定通道- conda-forge。
我在网上看到有人说用forge走的是外网,因此很慢导致加载卡住,因此,我把channels全部注释掉(默认没有channels项)然后测试,依然失败。
注:如果安装出现了Solving environment: failed with initial frozen solve. Retrying with flexible solve 错误
1 | conda update -n base conda |
即可解决
在后面的测试中,我发现metawrap和prokkaroary的安装
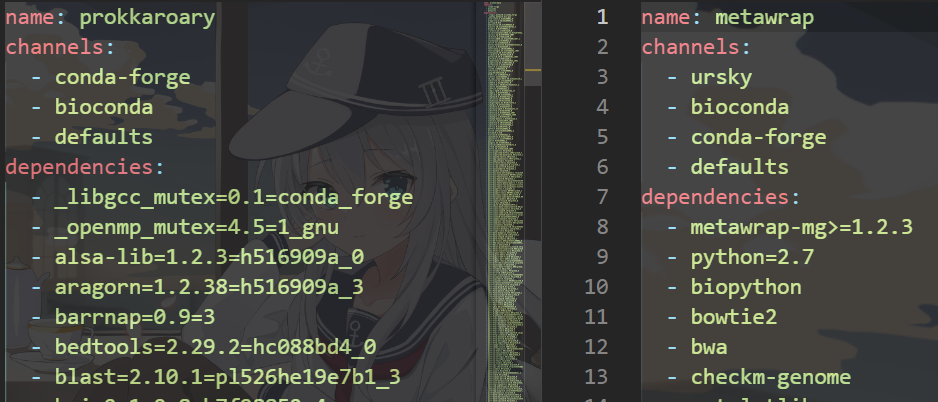
于是我们把后面的大于等于去掉在进行测试,等了一个小时也没有结果也失败了。
于是我们用土办法,创建环境然后一个一个手动输入加载
1 | conda create -p ./envs/metagem python=3.10 |
或者以下脚本
因为我每次下载都会报错JASON错误,网上一查是因为缓存的问题,因此我在每一个软件包安装之后会清理缓存,暂时我对这个问题没有很好的解决方法,如果没有这个问题的同学可以删除所有conda clean -i -y 语句,当然保留也没有任何问题。其次bioconda:: 这里面的库都是用bioconda 通道下载的,因此安装时要加上这个语句,不加的话很多包安装不上去。
1 | #conda create -p ./envs/metagem python=3.10 -y |
B.执行metaGEM.sh
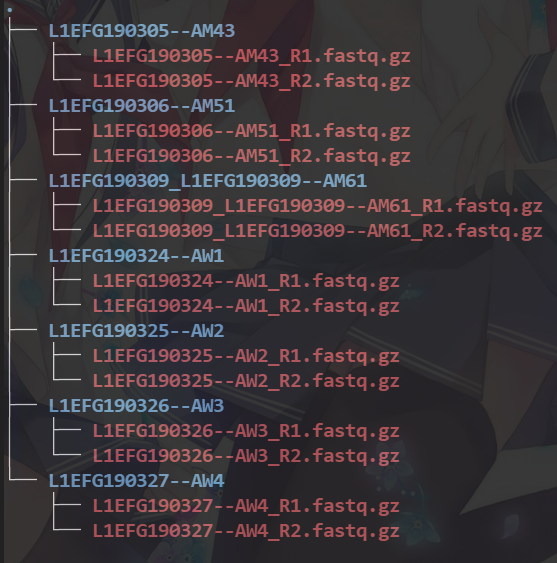
在dataset文件夹中的子目录中存放paierd-end的fastq数据,如下所示。MetGEM 将基于dataset文件夹中存在的子文件夹读取示例 ID,并将这些 ID 提供给 Snakefile 作为作业提交的通配符。
我的运行文件树如下图所示
1 | ├── colab |
使用fastp质量过滤reads
每个样本提交一个质量过滤工作,每个过滤工作有2个CPU和20GB 内存,最大运行时间为2小时
1 | bash metaGEM.sh -t fastp -j 2 -c 2 -m 20 -h 2 |
1.报错找不到路径
2024%201%EF%BC%88%E4%B8%80%EF%BC%89%202a55eec867d5441b9829bd5428024882/Untitled%206.png)
更改config/config.yaml 文件第一行的执行路径即可
如果还是找不到路径则在Snakefile中第一行更改config的路径
如果还是读取不到,则把config.yaml 文件复制到当前文件夹下
2.报错Error parsing number of cores (–cores, -c, -j): must be integer, empty, or ‘all’.
第一种情况snakemake版本过高,降低到5.10.0即可解决
第二种情况未执行pip install –user memote carveme smetana
3.报错找不到分析的数据
在snakefile文件中,我们可以看到第16/17行表示在dataset文件夹下,每一个文件名下面有两个同名加_R1,_R2的文件夹,因此我们要现将文件进行标准化操作,
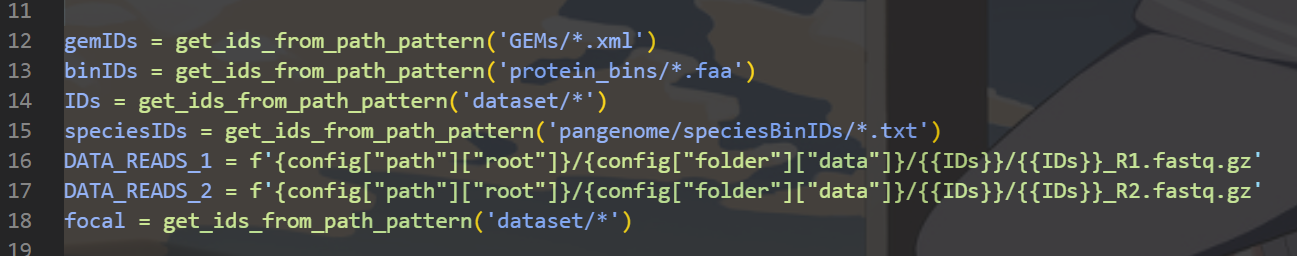
以下是我们的原始文件数据

我们要把他们进行分类,并且进行改名操作,因此我写了一个自动化分类脚本
1 | dataset_path="/home/gc/bash_all/0" #标准数据的文件夹 |
4.提交任务后,nohup.out提示sbatch: error: s_p_parse_file: unable to status file /etc/slurm-llnl/slurm.conf: No such file or directory, retrying in 1sec up to 60sec
(有的同学会提示/bin/sh: sbatch: command not found 之类的,这是没有安装slurm导致的看到有一篇文章写到需要这样安装https://www.thegeekdiary.com/sbatch-command-not-found/)
| Distribution | Command |
|---|---|
| Debian | apt-get install slurm-client |
| Ubuntu | apt-get install slurm-client |
| Kali Linux | apt-get install slurm-client |
| Fedora | dnf install slurm |
| OS X | brew install slurm |
| Raspbian | apt-get install slurm-client |
配置slurm有问题,slurm是一个linux服务器中的集群管理和作业调度系统,是项目里很关键的一点,因此要好好学习这里的配置信息
先看看文件
1 | ls -l /etc/slurm/ |
报错没有此文件,表明还没有安装slurm
弯路(后面还有未解决的报错)
(失败的教程)
https://blog.csdn.net/r1141207831/article/details/125272108
先从https://www.schedmd.com/这里下载,选择对应的版本
1
2
3
4
5
6
7
8
9
10#编译安装前需安装gcc
yum -y install gcc
#接着解压安装
tar -jxvf slurm-16.05.11.tar.bz2
cd /root/slurm-16.05.11
./configure
make
make install
#安装成功!在make和make install时出现
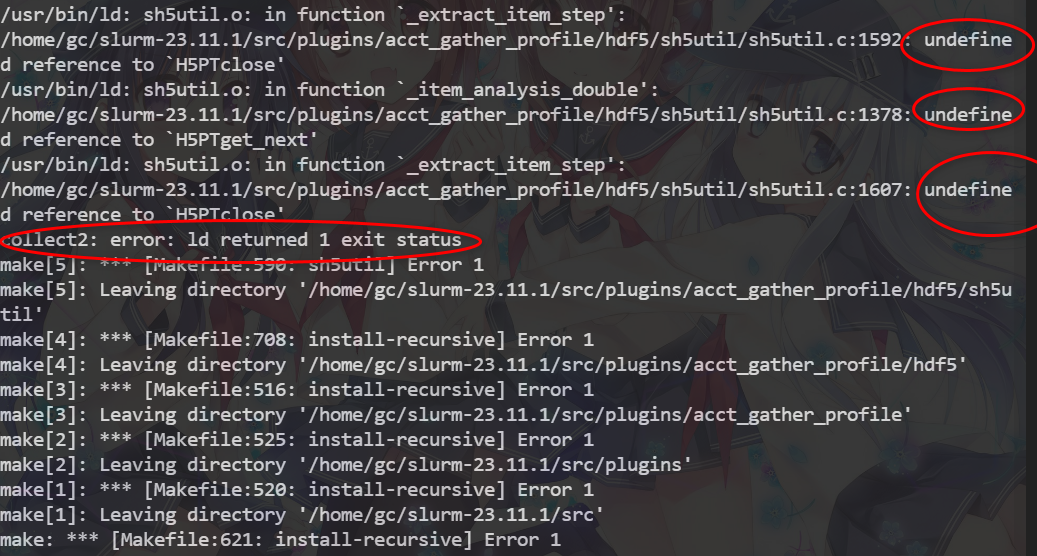
Ld 返回的1退出状态错误是以前错误的结果。有一个更早的错误ーー对‘hdf5各种方法的未定义引用造成的,因此我猜测是linux版本与slurm版本不同造成的,因此我找了一个适用于Ubuntu20.04 的slurm安装教程
最全slurm安装包列表如下https://src.fedoraproject.org/lookaside/extras/slurm/
a、安装必要文件
1
2sudo su
apt-get install make hwloc libhwloc-dev libmunge-dev libmunge2 munge mariadb-server libmysqlclient-dev -yb、启动启动munge服务
1
2
3systemctl enable munge // 设置munge开机自启动
systemctl start munge // 启动munge服务
systemctl status munge // 查看munge状态c、编译安装slurm
1
2
3
4
5
6
7# 将slurm-21.08.6.tar.bz2源码包放置在/home/fz/package目录下
cd /home/fz/package
tar -jxvf slurm-21.08.6.tar.bz2
cd slurm-21.08.6/
./configure --prefix=/opt/slurm/21.08.6 --sysconfdir=/opt/slurm/21.08.6/etc
make -j16
make install在make时发现缺少hdf5包
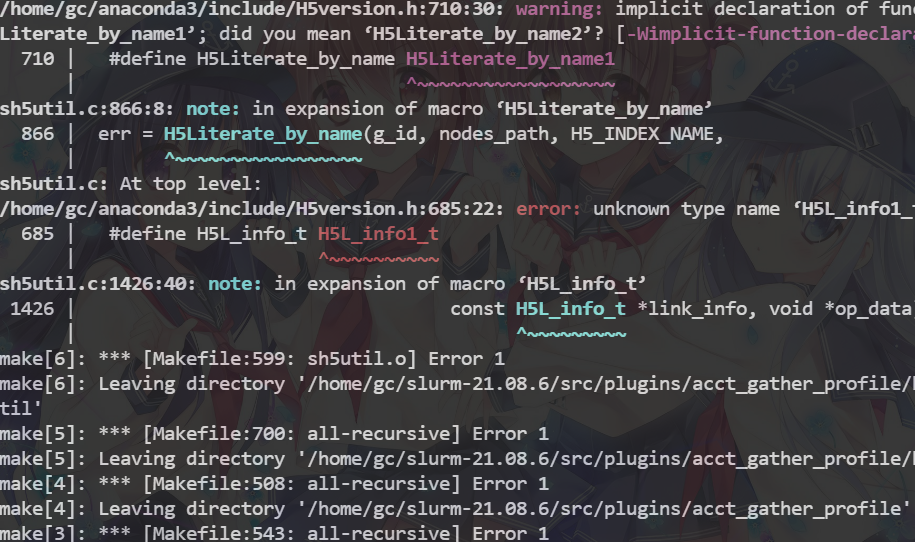
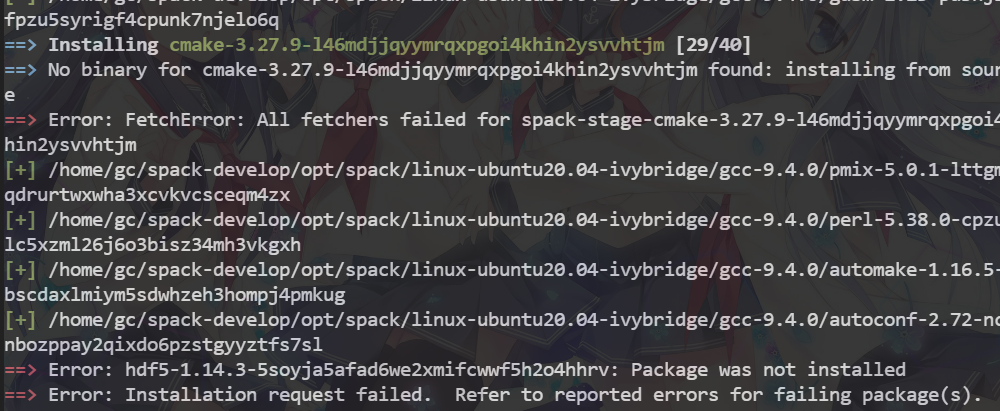
我又尝试使用spack高效的包管理器安装hdf5
https://hpc.pku.edu.cn/_book/guide/soft_env/spack.html(教程)
但是报错如下,只能进行手动下载编译
官网下载hdf5
https://support.hdfgroup.org/ftp/HDF5/releases/
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19sudo tar -xvf hdf5-1.8.21.tar.gz #执行解压
cd hdf5-1.8.21/ #sudo tar -xvf hdf5-1.8.21.tar.gz #执行解压
#依次执行
sudo ./configure --prefix=/usr/local/hdf5
sudo make #会有很多五颜六色的警告,忽略掉,
sudo make check
sudo make install
#安装成功后,在安装目录/usr/local下出现hdf5文件夹,打开后再切换到该目录下
cd /usr/local/hdf5/share/hdf5_examples/c
sudo ./run-c-ex.sh
#执行命令
sudo h5cc -o h5_extend h5_extend #如果显示错误,则安装:
sudo apt install hdf5-helpers
sudo apt-get install libhdf5-serial-dev
#再执行
sudo h5cc -o h5_extend h5_extend.c #直到执行后没有错误显示
#执行命令
sudo ./h5_extend1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19sudo tar -xvf hdf5-1.8.21.tar.gz #执行解压
cd hdf5-1.8.21/ #sudo tar -xvf hdf5-1.8.21.tar.gz #执行解压
#依次执行
sudo ./configure --prefix=/usr/local/hdf5
sudo make #会有很多五颜六色的警告,忽略掉,
sudo make check
sudo make install
#安装成功后,在安装目录/usr/local下出现hdf5文件夹,打开后再切换到该目录下
cd /usr/local/hdf5/share/hdf5_examples/c
sudo ./run-c-ex.sh
#执行命令
sudo h5cc -o h5_extend h5_extend #如果显示错误,则安装:
sudo apt install hdf5-helpers
sudo apt-get install libhdf5-serial-dev
#再执行
sudo h5cc -o h5_extend h5_extend.c #直到执行后没有错误显示
#执行命令
sudo ./h5_extend
装完hdf5后继续make slurm
没有报错!!!完成安装
完成安装,下面进行配置
d、启动数据库
后面是无限的hostname报错,我又重新找了一个教程,这个环境被污染了,如果使用另一个教程装slurm会冲突,但是这个又卸载不干净,于是重做了系统,在另一个教程上面成功了
正确的教程如下
来自https://wxyhgk.com/article%2Fubuntu-slurm(这个文章讲的太好了)
安装与配置
1 | #安装slurm |
配置slurm
配置文件是放在 /etc/slurm-llnl/ 下面的,使用命令
1 | sudo vi /etc/slurm-llnl/slurm.conf |
填写如下内容
1 | ClusterName=cool |
上面的代码中的
ControlMachine=master
PartitionName=master Nodes=master Default=NO MaxTime=INFINITE State=UP
#NodeName=master State=UNKNOWN
NodeName=master Sockets=2 CoresPerSocket=16 ThreadsPerCore=1 State=UNKNOWN
我 标红 和 标绿 的地方需要修改,这两部分是是需要修改的,其他的别动。
红色部分修改
使用
hostname命令可以查看到你的名字,然后把你的到的名字替换上面的 master绿色部分修改
这部分稍微有点复杂,首先来解释各个名字的意思
Sockets 代表你服务器cpu的个数
CoresPerSocket 代表每个cpu的核数
ThreadsPerCore 代表是否开启超线程,如果开启了超线程就是2,没有开启就是1
使用
vi [cxc.sh](http://cxc.sh/)写以下脚本1
2
3
4
5
6
7
8
9
10
11
12
13
14
cpunum=`cat /proc/cpuinfo| grep "physical id"| sort| uniq| wc -l`
echo "CPU 个数: $cpunum";
cpuhx=`cat /proc/cpuinfo | grep "cores" | uniq | awk -F":" '{print $2}'`
echo "CPU 核心数:$cpuhx" ;
cpuxc=`cat /proc/cpuinfo | grep "processor" | wc -l`
echo "CPU 线程数:$cpuxc" ;
if [[ `expr $cpunum\*$[cpuhx*2] ` -eq $cpuxc ]];
then
echo "开启了超线程"
else
echo "未开启超线程"
fi然后使用命令
bash [cxc.sh](http://cxc.sh/)运行脚本,看看线程数是不是核心数的两倍,如果是就开启了,没有就没开启。完成上面的之后吧对应的数字填写上去就可以了。
完成上述所有的设置之后就能启动服务了shell
1 | sudo systemctl enable slurmctld --now |
查看slurm队列信息
1 | sinfo |

如果这部分是 idle 就说明是可以的,如果不是 idle 请看这个
如果还是解决不了
比如是drain 其意思是用尽资源 解决文章
sinfo -R 报错Low socket***core***thre
那么直接把Sockets=2 CoresPerSocket=16 这两个参数减少,比如说除以2,留出一定的资源给系统使用,问题就解决了
确定目前队列里没有程序时,执行下列语句就好了(NodeName是上面设置的)
1 | scontrol update NodeName=m1 State=idle |
至此就已经安装完成了
到这里配置slurm就已经结束了
5.提交后sbatch: error: Batch job submission failed: No partition specified or system default partition
这个错误也是排查了好久,排查到这个文章
1 | [username@master1 ~]# sbatch example.sh --partition computeq #Note that ordering matters here! |
猜测是运行顺序错误导致的问题,于是我们到metaGEM.sh 中排查一下,核心的运行语句如下
1 | echo "nohup snakemake all -j $njobs -k --cluster-config ../config/cluster_config.json -c 'sbatch -A {cluster.account} -t {cluster.time} --mem {cluster.mem} -n {cluster.n} --ntasks {cluster.tasks} --cpus-per-task {cluster.n} --output {cluster.output}' &"|bash; break;; |
可以看到在后面的sbatch里面没有关于–partition的语句,于是我们手动添加,partition后面的名字就是前面我们设置的主机名
1 | --partition=你的主机名 |
1 | sbatch --partition=的主机名 -A {cluster.account} -t {cluster.time} --mem {cluster.mem} -n {cluster.n} --ntasks {cluster.tasks} --cpus-per-task {cluster.n} --output {cluster.output} |
代码中搜索sbatch 有三个地方需要添加,添加后即可正常运行
然后我们运行文章开头的语句
-j 任务数量
-c 每个任务CPU数量
-m 每个任务分配的内存大小
-h 每个任务运行的时间
注:注意CPU过大也不行
1 | bash metaGEM.sh -t fastp -j 5 -c 4 -m 20 -h 20 |
然后我们输入squeue 查看刚才提交的任务
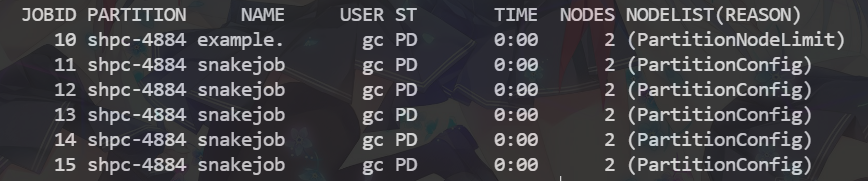
到这里我们的环境配置完毕